-
 Analytical & Elemental Instruments
Analytical & Elemental Instruments
-
Biotechnology Lab Items
-
Cardboard and Box Factory Lab Equipment Solution
-
Cleanroom Solution
-
Consumables & Accessories
-
Electrochemistry & Titration
-
ETP -Effluent Treatment Plant
-
Feed Quality Laboratory
-
Food & Kitchen Lab Items
-
Hygiene Testing Equipment
-
Lab Furniture
-
Laboratory Balances & Weighing Equipment
Laboratory Instrumentation in Modern Scientific Research: Classification, Functions, and Applications of Critical Analytical Instruments

Laboratories are the operational backbone of scientific investigation across disciplines spanning pharmaceutical research, clinical diagnostics, environmental monitoring, materials characterization, food safety analysis, and forensic science. The instruments housed within these facilities translate chemical, physical, and biological phenomena into quantitative data that supports hypothesis testing, product quality decisions, regulatory submissions, and public health protection.
The relationship between laboratory infrastructure and scientific output is direct and consequential. A research group’s capacity to generate reliable, reproducible, and regulatory-compliant data is bounded by the technical capabilities of its instrumentation, the validation status of its analytical methods, and the competency of its personnel. In pharmaceutical contexts particularly, where analytical data forms the evidential basis for drug registration, quality release, and patient safety determinations, the selection, qualification, and maintenance of laboratory instruments carries significance that extends beyond operational efficiency.
This article presents a structured technical examination of laboratory classification systems, the organization of pharmaceutical laboratory sections, the analytical methods employed across scientific disciplines, the operating principles and pharmaceutical significance of critical instruments, the regulatory framework governing laboratory operations in Bangladesh, and the challenges and prospects facing laboratory science as the pharmaceutical sector expands.
Classification of Laboratories
Classification by Scientific Discipline
Laboratories are organized and equipped according to the scientific disciplines and analytical objectives they serve. This functional differentiation determines facility design requirements, instrument selection, safety infrastructure, regulatory oversight frameworks, and personnel qualification standards.
Chemical Laboratories conduct synthesis, characterization, and quantitative analysis of chemical compounds using instruments including spectrophotometers, chromatography systems, titration apparatus, and elemental analyzers. These facilities range from basic teaching laboratories in educational institutions to sophisticated analytical chemistry laboratories in pharmaceutical manufacturing facilities performing validated pharmacopeial testing.
Biological and Microbiological Laboratories work with living organisms, cellular materials, nucleic acids, proteins, and microbial cultures. The biosafety requirements of these facilities depend on the risk classification of biological agents handled, ranging from Biosafety Level 1 for non-pathogenic organisms to Biosafety Level 4 for dangerous pathogens. Pharmaceutical microbiology laboratories performing sterility testing and bioburden determination represent a critical application of biological laboratory infrastructure in drug quality assurance.
Physical Testing Laboratories characterize the mechanical, thermal, optical, and electrical properties of materials using instruments including particle size analyzers, rheometers, thermal analysis systems, hardness testers, and dissolution apparatus. These facilities serve materials science research, pharmaceutical formulation development, and quality control testing of physical product attributes.
Environmental Laboratories analyze air, water, soil, and biological samples for contaminants, regulatory compliance parameters, and ecological health indicators. These facilities employ gas chromatography-mass spectrometry, inductively coupled plasma spectroscopy, electrochemical sensors, and biological assay systems for multi-matrix environmental analysis.
Clinical and Diagnostic Laboratories support medical decision-making through analysis of patient specimens including blood, urine, cerebrospinal fluid, and tissue sections. These facilities operate under healthcare-specific quality management frameworks including Clinical Laboratory Improvement Amendments in the United States and equivalent national standards in other jurisdictions.
Forensic Laboratories apply analytical science to legal investigation, performing toxicological analysis, document examination, trace evidence characterization, and DNA profiling using high-sensitivity instruments capable of working with limited and potentially degraded samples.
Classification by Function Within an Organization
Within industrial organizations including pharmaceutical manufacturers, laboratories fulfill distinct functional roles that reflect their position in the quality management and research hierarchy.
Quality Control Laboratories perform routine analytical testing of raw materials, in-process samples, and finished products against pharmacopeial or internal specifications to support batch release decisions. These facilities require validated methods, calibrated instruments, and comprehensive documentation systems meeting GMP requirements.
Quality Assurance Laboratories focus on system oversight, method validation, instrument qualification management, and regulatory compliance activities rather than primary sample analysis. Personnel in these sections review and approve quality control data, manage deviation and change control processes, and prepare documentation supporting regulatory submissions.
Research and Development Laboratories operate with greater analytical flexibility, employing exploratory techniques and developing new methods to characterize drug candidates, investigate formulation variables, and establish the analytical foundation for future product registrations. These facilities typically house a broader range of instrumentation and tolerate higher analytical uncertainty during early development phases.
Stability Testing Laboratories conduct formal degradation studies under controlled environmental conditions to establish product shelf life and support storage recommendations in regulatory submissions. These facilities require validated environmental chambers, calibrated monitoring systems, and documentation frameworks meeting ICH Q1A guideline requirements.
Structure and Workflow of a Pharmaceutical Laboratory
Physical Design and Environmental Controls
The architectural organization of a pharmaceutical laboratory reflects both functional workflow requirements and regulatory compliance obligations. GMP guidelines specify physical separation between areas performing incompatible activities, adequate space for instrument operation and maintenance, appropriate ventilation and air handling systems, and surfaces that support cleaning and decontamination procedures.
Laboratory design in GMP-compliant pharmaceutical facilities separates sample receipt and preparation areas from instrument analysis zones to prevent cross-contamination. Reference standard storage occurs in dedicated, access-controlled areas with temperature and humidity monitoring. Glassware washing and preparation facilities occupy separate spaces from analytical areas to prevent contamination of clean equipment. Chemical storage follows compatibility groupings and ventilation requirements that prevent hazardous atmospheric accumulations.
Environmental monitoring programs in pharmaceutical quality control laboratories track temperature, relative humidity, differential pressure between zones, and particulate contamination levels against defined specifications. Deviations from specified ranges trigger documented investigations and, where necessary, reassessment of analytical results generated during affected periods.
Quality Control Section Operations
The quality control section of a pharmaceutical laboratory represents the primary analytical testing function within the quality management system. Chemists and analysts in this section execute validated test procedures against approved specifications, generating primary data that directly supports batch disposition decisions.
Standard operating procedures govern every aspect of quality control operations including instrument startup and shutdown sequences, system suitability testing requirements before analytical runs, sample preparation protocols, calculation methods, acceptance criteria application, and out-of-specification investigation procedures. These procedures, maintained under document control and subject to periodic review, ensure that analytical operations remain consistent regardless of which qualified analyst performs the work.
Workload management in quality control sections requires careful scheduling of instrument time across competing testing requirements from incoming material testing, in-process monitoring, finished product release, stability sample analysis, and method validation activities. Chromatographic systems capable of unattended overnight operation through autosampler configurations extend effective instrument utilization beyond staffed hours, increasing throughput without proportional increases in personnel requirements.
Research and Development Laboratory Organization
Pharmaceutical research and development laboratories support preformulation characterization, formulation optimization, process development, and analytical method development activities that precede formal regulatory submissions. These sections operate under somewhat different documentation and procedural standards than quality control laboratories, reflecting the exploratory nature of early development work.
Preformulation studies in R&D laboratories characterize drug substance physical and chemical properties including solubility across pH ranges, pKa values, polymorphic forms, hygroscopicity, photostability, and degradation pathways. This fundamental characterization data guides formulation strategy selection and identifies analytical challenges that must be addressed during method development. Instruments deployed in preformulation characterization include differential scanning calorimeters, X-ray powder diffraction systems, dynamic vapor sorption analyzers, and dissolution testing equipment alongside conventional chromatographic and spectroscopic tools.
Method development activities progress from initial method scouting using generic column and mobile phase conditions through systematic optimization of resolution, sensitivity, and specificity parameters to formal validation studies demonstrating compliance with ICH Q2(R1) requirements. Each development stage generates documentation that traces the scientific rationale for analytical decisions and provides the knowledge base supporting future method transfers and troubleshooting.
Microbiology Section
The pharmaceutical microbiology section performs testing critical to product safety for parenteral, ophthalmic, and other sterile dosage forms, as well as non-sterile products with defined microbial quality limits. Core testing activities include sterility testing of finished sterile products, bioburden determination of non-sterile products and in-process materials, endotoxin testing using limulus amebocyte lysate or recombinant Factor C methods, and antimicrobial effectiveness testing of preserved products.
Environmental monitoring programs operated by the microbiology section characterize the microbiological quality of manufacturing and laboratory cleanroom environments through active air sampling, passive settle plate exposure, surface contact sampling, and personnel monitoring. Trending of environmental monitoring data provides early warning of deteriorating contamination control conditions before product quality is affected.
Analytical Methods in Scientific Laboratories
Spectroscopic Methods
Spectroscopic analytical methods characterize and quantify substances through measurement of electromagnetic radiation interactions with matter. These interactions span the full electromagnetic spectrum and provide structural information, identity confirmation, and quantitative concentration data across diverse sample types and matrices.
Ultraviolet and visible spectrophotometry exploits the absorption of radiation in the 190 to 800 nanometer range by electronic transitions in organic chromophores and coordination complexes. The Beer-Lambert relationship provides the theoretical basis for quantitative analysis, relating measured absorbance directly to analyte concentration through the product of molar absorptivity and optical path length. Applications in pharmaceutical laboratories include drug assay in formulations, dissolution profile monitoring, protein quantification, and color measurement.
Infrared spectroscopy characterizes molecular structure through measurement of vibrational transitions occurring when molecules absorb infrared radiation at frequencies corresponding to specific bond stretching and bending modes. Attenuated total reflectance Fourier transform infrared spectroscopy has become the dominant implementation of infrared analysis in pharmaceutical laboratories, providing rapid, non-destructive identification of drug substances, excipients, and solid dosage forms through comparison of measured spectra against reference databases.
Nuclear magnetic resonance spectroscopy provides detailed structural information about molecular frameworks through measurement of radio-frequency energy absorption by atomic nuclei with non-zero spin quantum numbers when placed in strong magnetic fields. Proton and carbon-13 NMR spectroscopy are routinely used for structural confirmation of synthesized compounds, purity assessment of reference standards, and polymorph identification in pharmaceutical development.
Atomic absorption spectroscopy and inductively coupled plasma spectroscopy determine elemental composition by measuring the absorption or emission of radiation by atoms in the gas phase. These techniques are applied to elemental impurity analysis in pharmaceutical drug substances and finished products per ICH Q3D guideline specifications.
Chromatographic Methods
Chromatographic separation methods resolve mixtures into individual components based on differential migration through a stationary phase under the influence of a mobile phase. The resulting separation enables identification and quantification of individual components in complex samples that would be analytically intractable without prior separation.
High-performance liquid chromatography separates analytes dissolved in mobile phase solutions through differential partitioning between mobile and stationary phases under high pressure. Reversed-phase chromatography on octadecylsilyl-bonded silica stationary phases represents the most widely employed mode, separating analytes based primarily on hydrophobic interactions. Ion-exchange, normal phase, and size exclusion chromatography modes extend the technique to ionic compounds, non-polar matrices, and macromolecular analytes respectively.
Gas chromatography separates volatile and semi-volatile compounds by partitioning between a carrier gas mobile phase and a liquid or solid stationary phase coated on capillary column walls. Temperature programming allows sequential elution of compounds across wide volatility ranges within a single analytical run. Gas chromatography hyphenated with mass spectrometry provides definitive structural confirmation alongside quantitative analysis, making GC-MS a standard technique for residual solvent identification, volatile impurity characterization, and forensic analysis.
Electrochemical Methods
Electrochemical analytical methods measure electrical quantities arising from chemical reactions at electrode-solution interfaces. Potentiometric methods measure cell potentials under zero-current conditions using ion-selective electrodes, with the pH glass electrode representing the most widely deployed example. Amperometric and voltammetric methods apply controlled potentials to working electrodes and measure resulting current flows as functions of potential or time, providing concentration-proportional analytical signals for electroactive analytes.
Karl Fischer titration, a specialized coulometric or volumetric method for water content determination, operates through quantitative reaction of water with iodine in the presence of sulfur dioxide and a base. This method provides the most reliable and specific determination of water content in pharmaceutical materials, directly relevant to stability and processing performance of moisture-sensitive formulations.
Sample Preparation and Method Validation
Analytical method performance depends critically on sample preparation procedures that extract, concentrate, and clarify analytes from complex matrices before instrumental measurement. Solid-liquid extraction using appropriate solvents, protein precipitation for biological samples, solid-phase extraction for trace enrichment, and microwave-assisted digestion for elemental analysis represent the principal sample preparation approaches in pharmaceutical analytical laboratories.
Method validation according to ICH Q2(R1) demonstrates that an analytical procedure is fit for its intended purpose through systematic evaluation of specificity, linearity, range, accuracy, precision, detection limit, quantification limit, and robustness parameters. Each validation parameter requires defined experimental designs, appropriate statistical analysis of results, and comparison against acceptance criteria that reflect the analytical purpose and regulatory requirements.
Key Laboratory Instruments: Principles and Applications
UV-Vis Spectrophotometer
The ultraviolet-visible spectrophotometer measures the intensity of electromagnetic radiation transmitted through or reflected from a sample as a function of wavelength across the ultraviolet and visible spectral regions. The instrument comprises a radiation source, wavelength selection device, sample compartment, and detector system. Deuterium arc lamps provide ultraviolet radiation from approximately 190 to 400 nanometers, while tungsten-halogen lamps cover the visible range from 350 to 800 nanometers. Modern instruments switch automatically between sources at defined wavelengths.
Wavelength selection in single-beam and double-beam designs employs diffraction gratings that disperse polychromatic radiation into its spectral components, with a variable exit slit selecting the desired wavelength band for transmission to the sample. Photodiode array detectors used in diode array spectrophotometers collect the complete spectrum simultaneously by dispersing transmitted radiation onto a linear detector array, eliminating the need for wavelength scanning and enabling rapid multi-wavelength data acquisition.
Double-beam instrument architectures split the source radiation into reference and sample beams that pass through matched reference and sample cells respectively, with the detector comparing the two beam intensities to generate absorbance values. This configuration cancels source intensity fluctuations and reduces the effect of solvent absorption on analytical measurements. Pharmaceutical applications include drug substance assay by direct absorbance measurement, determination of dissolution profiles at defined sampling time points, and identification testing by comparison of absorption maxima with pharmacopeial reference spectra.
High-Performance Liquid Chromatography
High-performance liquid chromatography systems consist of a solvent delivery module providing precise, pulseless mobile phase flow at pressures reaching several hundred bar, a sample injection system introducing defined volumes of prepared samples into the mobile phase stream, an analytical column containing the stationary phase, a detector measuring analyte concentration in column effluent, and a data system processing detector signals into chromatographic data.
The separation mechanism in reversed-phase HPLC operates through hydrophobic interactions between analyte molecules and the nonpolar octadecylsilyl stationary phase surface. Analytes with greater hydrophobicity partition more strongly into the stationary phase and elute later than more polar compounds. Mobile phase composition, typically a mixture of water with acetonitrile or methanol, controls the strength of hydrophobic interactions and therefore analyte retention times. Gradient elution progressively increases organic modifier concentration during the analytical run to elute compounds across a wide polarity range.
Ultraviolet absorbance detection at fixed or variable wavelengths provides sensitive detection for compounds with appropriate chromophores, while photodiode array detection records complete ultraviolet-visible spectra at each chromatographic time point, enabling spectral purity assessment and identity confirmation of resolved peaks. Fluorescence detection offers enhanced sensitivity for fluorescent compounds, and mass spectrometric detection provides structural information and ultra-trace sensitivity for targeted and untargeted analyses.
Pharmaceutical HPLC applications include assay of drug substances and drug products, related substance and degradation product profiling, dissolution testing sample analysis, stability-indicating method development, and cleaning validation residue determination. System suitability requirements specified in pharmacopeial methods verify column efficiency, peak symmetry, resolution between critical peak pairs, and injection repeatability before each analytical sequence.
Gas Chromatography Systems
Gas chromatographic systems operate by injecting liquid or gaseous samples into a heated injection port where volatile analytes vaporize into the carrier gas stream. The carrier gas, typically helium, hydrogen, or nitrogen at flow rates between 1 and 5 milliliters per minute, transports vaporized analytes through a fused silica capillary column with internal diameters between 0.1 and 0.53 millimeters coated with stationary phase films of defined polarity and thickness.
The column is housed within a temperature-programmable oven that maintains isothermal conditions for simple applications or ramps temperature from an initial value through one or more programmed stages to accelerate elution of higher-boiling components. Stationary phase polarity determines separation selectivity; nonpolar phases retain nonpolar analytes preferentially through dispersion interactions, while polar phases interact with polar analytes through dipolar and hydrogen bonding mechanisms.
Flame ionization detectors ionize organic molecules in a hydrogen-air diffusion flame, producing ion currents proportional to the mass flow rate of carbon-containing compounds entering the detector. This near-universal response to organic compounds combined with wide linear dynamic range makes flame ionization detection the standard choice for quantitative organic analysis including residual solvent determination. Thermal conductivity detectors respond to differences in thermal conductivity between carrier gas and analyte molecules, providing universal detection applicable to inorganic gases and compounds without measurable flame ionization response.
In pharmaceutical quality control laboratories in Bangladesh, gas chromatography performs residual solvent testing per ICH Q3C and applicable pharmacopeial methods, volatile impurity profiling in drug substances, headspace analysis of container closure systems, and identity testing of specific volatile active pharmaceutical ingredients. System suitability demonstrations before each analytical sequence verify retention time reproducibility, detector sensitivity, peak symmetry, and resolution between critical solvent pairs.
pH Meters and Potentiometric Instruments
The measurement of hydrogen ion activity by glass combination electrodes provides one of the most fundamental measurements in pharmaceutical analysis, affecting dissolution medium preparation, buffer solution quality verification, stability study sample conditioning, and formulation development investigations. The glass electrode generates a potential at its membrane surface governed by the Nernst equation, with each unit change in pH producing approximately 59.16 millivolts of potential change at 25 degrees Celsius.
Modern bench-top pH meters employ microprocessor-controlled measurement algorithms that apply temperature compensation corrections to electrode readings either through automatic temperature electrode immersion in the sample or through manual temperature entry. Multi-point calibration using two or three certified buffer standards spanning the measurement range corrects for electrode slope and offset deviations from theoretical Nernst behavior.
Ion-selective electrode measurement systems extend potentiometric analysis beyond pH to include fluoride, nitrate, ammonium, chloride, sodium, calcium, and other ionic species. These measurements support water quality assessment in pharmaceutical utility systems, raw material identity testing, and investigation of ionic species affecting drug stability or bioavailability.
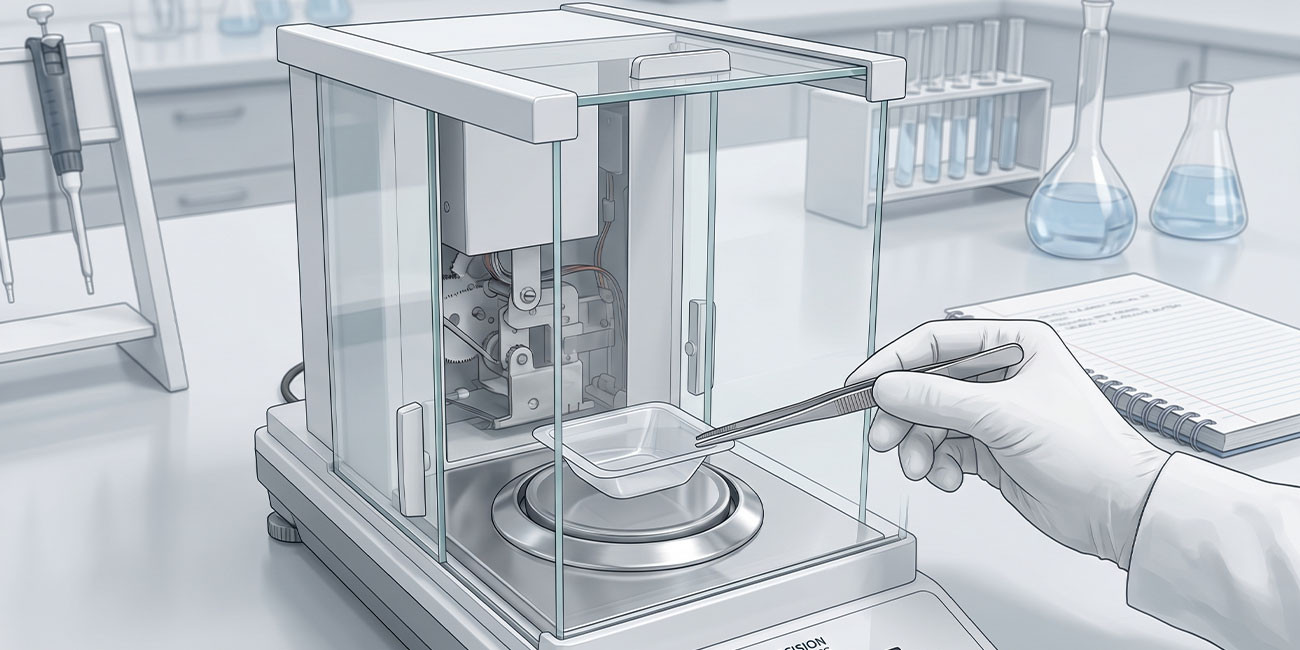
Analytical Balances
Analytical balances based on electromagnetic force restoration technology determine sample mass by generating an electromagnetic force sufficient to maintain the balance pan at a fixed position against the gravitational force exerted by the sample. The magnitude of the electrical current required to maintain positional equilibrium provides a signal proportional to sample mass, which is processed and displayed after calibration against reference weights.
Modern analytical balances achieve readabilities of 0.01 to 0.1 milligrams with linearity specifications within plus or minus 0.2 to 0.5 milligrams across working ranges typically spanning 0 to 200 grams. Temperature sensitivity, vibration susceptibility, and air current effects represent the primary environmental factors limiting measurement precision. Draft shields enclosing the weighing pan reduce air current disturbances, while anti-vibration tables support balances in laboratories where floor vibrations from building services or nearby equipment create measurement instability.
Calibration programs for analytical balances in pharmaceutical GMP laboratories employ certified reference weights traceable to national or international mass standards through unbroken calibration chains. External calibration using reference weights at multiple points across the balance working range verifies linearity and accuracy. Automated internal calibration systems available in many modern balances use built-in reference weights to apply correction factors continuously or on demand without operator intervention.
Weighing accuracy in pharmaceutical quality control laboratories directly affects the accuracy of all subsequent analytical measurements because reference standard preparation, sample solution preparation, and reagent formulation all depend on accurate mass determination. Systematic errors in weighing propagate through analytical calculations and, if unrecognized, produce biased assay results with regulatory and patient safety implications.
Particle Size Analyzers
Pharmaceutical particle size analysis characterizes size distributions of drug substance crystals, inhalation aerosol droplets, emulsion globules, suspension particles, and granulation materials. The measured size distribution influences dissolution rate, aerodynamic behavior of inhaled particles, biological membrane permeability, physical stability of suspensions and emulsions, and compaction behavior during tablet manufacturing.
Laser diffraction particle size analyzers measure the angular distribution of laser light scattered by particles dispersed in liquid or aerosol form. Particles of different sizes scatter light at characteristic angles with intensities governed by Mie scattering theory for particles comparable in size to the laser wavelength, and by Fraunhofer diffraction theory for larger particles where full Mie theory is unnecessary. The scattered light pattern recorded by a semicircular detector array is mathematically inverted to calculate the volume-weighted particle size distribution that best reproduces the observed scattering pattern.
Dynamic light scattering instruments measure the time-dependent fluctuations in scattered light intensity caused by Brownian motion of particles in suspension. These fluctuations occur at rates inversely related to particle size through the Stokes-Einstein equation relating diffusion coefficient to hydrodynamic diameter. Dynamic light scattering provides high-sensitivity characterization of nanoparticles, liposomes, protein aggregates, and other submicron species in the 1 nanometer to 10 micrometer size range relevant to advanced drug delivery systems.
Aerodynamic particle sizers and impactor systems characterize inhalation drug products by measuring particle aerodynamic behavior rather than geometric size alone, providing data directly relevant to pulmonary deposition prediction. Cascade impactors separate aerosol particles into size fractions based on inertial impaction, generating deposition profiles across stages representing different respiratory tract deposition sites.
Regulatory Framework for Pharmaceutical Laboratories in Bangladesh
Directorate General of Drug Administration
The Directorate General of Drug Administration functions as the primary pharmaceutical regulatory authority in Bangladesh, established under the Ministry of Health and Family Welfare. This organization regulates the registration, licensing, manufacturing, import, export, distribution, and quality surveillance of pharmaceutical products within Bangladesh.
The DGDA conducts GMP inspections of pharmaceutical manufacturing facilities against standards aligned with World Health Organization guidelines and Pharmaceutical Inspection Co-operation Scheme technical documents. These inspections evaluate manufacturing facility design, equipment qualification status, analytical laboratory capabilities, quality management systems, personnel qualification programs, and documentation controls. Inspection outcomes determine manufacturing license renewal decisions and can restrict production or distribution of specific products where serious compliance deficiencies are identified.
The regulatory authority has progressively tightened compliance expectations over recent years, reflecting both domestic quality improvement objectives and the requirements of export market regulatory authorities in Europe, North America, and regulated Asian markets. Pharmaceutical companies seeking to maintain or expand export market access must demonstrate compliance standards meeting the expectations of foreign regulatory authorities, which in some cases exceed DGDA baseline requirements.
Good Manufacturing Practice Requirements
GMP guidelines applicable to pharmaceutical laboratories in Bangladesh specify comprehensive requirements across analytical operations that collectively ensure the reliability and integrity of data supporting product quality decisions. These requirements address facility and equipment standards, qualification and calibration programs, analytical method validation, reference standard management, reagent and solution preparation, sample management, and electronic data integrity.
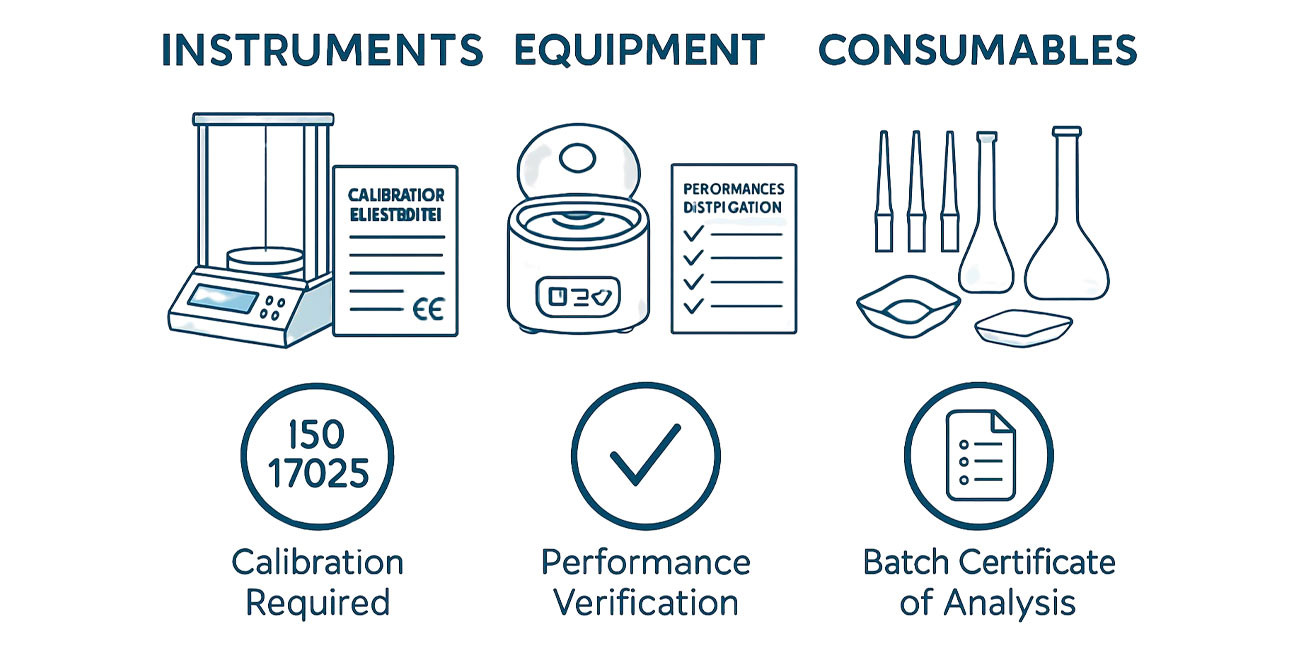
Equipment qualification under GMP frameworks proceeds through documented Installation Qualification, Operational Qualification, and Performance Qualification phases that collectively demonstrate instruments are correctly installed, operate within manufacturer specifications, and consistently produce accurate results when used for defined analytical purposes. Installation Qualification verifies that equipment specifications match procurement requirements and that utility connections, environmental conditions, and spatial arrangements comply with requirements. Operational Qualification testing verifies performance across the instrument’s working range using appropriate reference standards and traceable measurement equipment. Performance Qualification demonstrates fitness for purpose in specific analytical procedures through studies using representative sample types and processing conditions.
Calibration management programs maintain metrological traceability for measurements performed by laboratory instruments. Calibration certificates document measurement values, uncertainties, acceptance criteria, pass or fail determinations, and reference measurement equipment used, providing documented evidence that instrument measurements remain accurate and within specification throughout the calibration period.
Data integrity requirements in GMP environments address the complete lifecycle of analytical data from initial generation through long-term archival. ALCOA-plus principles — requiring that data be attributable, legible, contemporaneous, original, accurate, complete, consistent, enduring, and available — govern data generation, recording, processing, review, and storage practices. Computerized laboratory systems require validation demonstrating that software functions correctly, access controls prevent unauthorized data modification, and audit trails provide complete records of all data creation and modification activities.
International Standards and Pharmacopeial Compliance
The United States Pharmacopeia, British Pharmacopeia, and European Pharmacopeia each contain analytical procedures and specifications forming the basis for quality testing of pharmaceutical products manufactured in Bangladesh for both domestic and export markets. While the British Pharmacopeia has historically predominated in Bangladesh due to the country’s Commonwealth connections, manufacturers targeting United States markets must implement analytical methods meeting United States Pharmacopeia specifications, which sometimes differ in technical detail from British Pharmacopeia counterparts.
WHO prequalification programs provide a pathway for pharmaceutical manufacturers in developing countries to demonstrate product quality meeting international standards acceptable for procurement by United Nations agencies and regulated national health programs. Achieving WHO prequalification requires documented demonstration that manufacturing facilities, quality management systems, and analytical testing capabilities meet WHO GMP standards, creating an internationally recognized quality credential that facilitates market access in developing country markets.
ISO 17025 laboratory accreditation through national accreditation bodies provides independent technical assessment of laboratory competence, calibration management, quality system implementation, and measurement uncertainty management. Laboratories achieving ISO 17025 accreditation demonstrate technical capabilities meeting this international standard through rigorous assessments including proficiency testing participation and on-site evaluation by trained assessors.
The Role of Pharmaceutical Laboratories in Bangladesh
Drug Safety and Patient Protection
The analytical testing performed by pharmaceutical quality control laboratories in Bangladesh represents the primary technical defense against the distribution of substandard, degraded, or mislabeled pharmaceutical products to patients. Identity testing confirms that materials received from suppliers contain the declared active pharmaceutical ingredients and excipients. Purity testing detects potentially harmful impurities arising from synthesis, degradation, or contamination. Assay testing verifies that drug products contain declared quantities of active ingredients within acceptable tolerances that ensure therapeutic effectiveness.
The domestic pharmaceutical market in Bangladesh reached approximately USD 3.37 billion by mid-2025 according to industry reports, reflecting continued expansion driven by population growth, increasing healthcare access, and the introduction of new therapeutic categories. The volume of drug products manufactured for this market correspondingly increases the analytical testing burden carried by quality control laboratories and the public health consequences of analytical failures.
The export of pharmaceutical products to more than 100 countries generates additional quality assurance requirements because products failing quality standards in destination countries damage the reputation not only of individual manufacturers but of Bangladesh’s pharmaceutical export sector as a whole. Documented analytical failures resulting in product recalls or import alerts create direct commercial losses and long-term market access complications.
Supporting Industrial Development and Economic Growth
The pharmaceutical sector contributes significantly to Bangladesh’s economy through direct employment, export revenue generation, and the development of technical human capital. Quality control and analytical laboratory operations within this sector employ analytical chemists, pharmacists, microbiologists, and quality assurance specialists whose technical skills represent transferable capabilities supporting broad scientific and industrial development.
Investment in modern laboratory instrumentation and supporting analytical infrastructure increases the technical sophistication of pharmaceutical companies’ quality systems, enabling them to compete for more complex manufacturing contracts, penetrate regulated export markets with high quality standards, and develop more technically advanced product categories including biosimilars, controlled-release formulations, and sterile injectable products.
The development of active pharmaceutical ingredient manufacturing capability in Bangladesh, supported by dedicated API industrial parks, creates additional demand for sophisticated analytical laboratory capabilities appropriate for characterizing synthetic intermediates, monitoring chemical processes, and verifying API quality against strict purity specifications.
Research Capacity and Innovation
Pharmaceutical research and development laboratories in Bangladesh represent an area of relative underdevelopment compared to the manufacturing sector’s maturity. The majority of pharmaceutical companies in Bangladesh have historically focused on generic product manufacturing with limited investment in novel drug research. However, changing competitive dynamics, including the eventual loss of least-developed country trade advantages following LDC graduation, are creating incentives for increased investment in formulation innovation and analytical capability development.
Strengthening analytical laboratory infrastructure in both industry and academic research institutions creates the foundation for expanded pharmaceutical research activities, improved product formulation capabilities, and enhanced capacity to generate the analytical data supporting regulatory submissions for new product registrations in domestic and international markets.
Challenges in Laboratory Operations and Instrumentation
Infrastructure and Technical Limitations
Pharmaceutical laboratories in Bangladesh face infrastructure challenges that affect both analytical performance and operational continuity. Electrical power supply instability in some locations requires investment in uninterruptible power supply systems and voltage conditioning equipment to protect sensitive analytical instruments from power quality issues that can corrupt analytical runs, damage electronic components, or compromise data integrity.
High-purity water requirements for HPLC mobile phase preparation, dissolution medium preparation, and reagent solution preparation demand reliable water purification infrastructure meeting Type I water specifications. Variability in municipal water supply quality creates challenges for water purification system performance and necessitates regular monitoring of purified water quality parameters.
Tropical climate conditions with elevated ambient temperatures and humidity levels affect analytical instrument performance, reagent stability, and reference standard integrity. Adequate air conditioning in all laboratory areas is not merely a comfort consideration but a technical requirement for maintaining instruments within their specified operating temperature ranges and preventing humidity-related degradation of moisture-sensitive materials.
Human Resource Development
The expansion of pharmaceutical manufacturing capacity in Bangladesh has created sustained demand for qualified laboratory personnel that exceeds the output of relevant educational programs. Graduate chemists and pharmacists entering laboratory positions frequently require substantial additional training in GMP procedures, analytical instrument operation, and quality system documentation before they can independently perform validated analytical procedures.
Structured competency development programs that combine documented training activities, supervised practical work, and formal assessment represent the systematic approach to personnel development that GMP requirements and quality management best practices demand. However, the implementation quality of such programs varies substantially across organizations, creating inconsistencies in analytical workforce competency.
Retention of experienced analytical personnel is an ongoing challenge in competitive pharmaceutical labor markets. The loss of qualified analysts through turnover requires repeated training investments and can temporarily reduce laboratory throughput and procedural consistency during transition periods.
Data Integrity and Computerized System Validation
Data integrity management in pharmaceutical analytical laboratories has become a major focus of regulatory inspections globally, with inspectors from multiple regulatory authorities identifying data integrity deficiencies as a leading source of compliance observations and enforcement actions. The migration from paper-based to electronic analytical records systems, while offering many operational advantages, introduces data governance requirements including system validation, access control management, audit trail configuration, and backup procedures.
Computerized system validation for analytical data acquisition systems including chromatography data systems, spectrophotometer software, and laboratory information management systems requires documented evidence that software functions correctly, that data cannot be modified without detection through audit trail records, and that backup systems reliably preserve analytical data against loss. These validation requirements represent significant resource investments but are non-negotiable for laboratories operating under GMP frameworks targeting regulated market access.
Technological Advances and Future Directions
Automation and High-Throughput Analysis
Laboratory automation technologies are changing pharmaceutical analytical operations through integration of robotic liquid handling, automated sample preparation, and computerized workflow management with analytical instrument networks. These systems reduce manual handling variability, increase analytical throughput, and free analytical scientists from repetitive sample preparation tasks to focus on data interpretation, method development, and quality oversight activities.
Automated dissolution testing systems with robotic sampling, in-line filtration, and direct HPLC injection eliminate multiple manual handling steps in traditional dissolution analysis workflows. These systems improve data quality through reduced analyst-to-analyst variability in sampling timing and filtration technique, while simultaneously increasing throughput for formulation screening and quality control applications.
Hyphenated and Multi-Dimensional Analytical Techniques
Hyphenated instruments combining separation techniques with structural characterization detectors have become standard tools in advanced pharmaceutical analytical laboratories. Liquid chromatography-mass spectrometry systems provide simultaneous separation, quantification, and structural confirmation capabilities that individual LC or MS instruments cannot deliver separately. Ultra-high-performance liquid chromatography systems using sub-2-micrometer particle columns achieve faster separations with higher resolution and reduced solvent consumption compared to conventional HPLC. Two-dimensional chromatography systems address complex matrix separations that exceed the peak capacity of single-column analyses.
Artificial Intelligence and Computational Analytics
Machine learning algorithms applied to spectroscopic and chromatographic data streams provide enhanced capabilities for impurity characterization, method development acceleration, and equipment performance monitoring. Chemometric modeling of spectral datasets enables quantitative prediction of multiple analyte concentrations from single spectroscopic measurements, reducing analytical run times compared to sequential single-analyte methods.
Predictive maintenance algorithms analyzing instrument performance data identify degradation patterns before they produce analytical failures, enabling proactive maintenance scheduling that prevents unplanned instrument downtime. These capabilities hold particular value in pharmaceutical quality control operations where instrument availability directly affects production scheduling and batch release timelines.
For pharmaceutical laboratories in Bangladesh navigating increasing analytical complexity alongside regulatory compliance demands, these technological developments represent both opportunities and challenges. Organizations that strategically invest in upgraded instrumentation, validated computerized systems, and technically qualified personnel create the analytical capability foundation necessary for sustained competitiveness in domestic and international pharmaceutical markets.
Recent Posts


Gas Analysis and Monitoring: Principles of Multi-Gas Detection and Environmental Applications

Spectroscopic Techniques in Chemical Analysis: Principles, Types, and Laboratory Applications

Laboratory Mixing and Shaking Systems: Mechanical Principles and Applications in Scientific Research


Sterility Testing and Contamination Control in Pharmaceutical and Microbiological Laboratories
Optimum Solution (OS4U) – Laboratory & Analytical Instruments Supplier in Bangladesh
Optimum Solution (OS4U) is a leading supplier of laboratory equipment, analytical instruments, and process control solutions in Bangladesh. Since our inception, we have built a strong presence in the industry, serving research labs, universities, pharmaceutical companies, and quality control laboratories nationwide. Through continuous dedication to improving our products and services, Optimum Solution has earned the trust and loyalty of our valued clients.
